 In Organic Chemistry, synthesis and retrosynthesis go hand in hand.
In Organic Chemistry, synthesis and retrosynthesis go hand in hand.
While there isn’t a clear distinction, I like to think of synthesis as forward thinking and retrosynthesis as the reverse.
Synthesis is a topic that is typically introduced in Organic Chemistry 1, right after studying alkyne reactions. You’ll be utilizing it again and again as the number of reactions that you learn accumulate.
This is why it is important to review past topics prior to moving on to the next chapter. While learning the new topics, you may be asked to perform a retrosynthesis that involves retrieving five different reactions from five different chapters.
Let’s back up…
What is retrosynthesis?
Retro = Backwards
Synthesis = The process of combining simpler reactions to form a chemical compound/molecule.
In your Organic Chemistry course, this is presented in the form of a complex molecule that you are then asked to synthesize from a given starting molecule, or a set of reaction conditions.
–>You may be given a specific reactant and asked to synthesize a product.
For example:
“Synthesize trans 3,4-dibromohexane from ethyne.”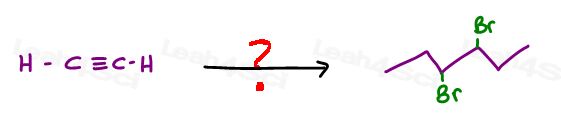
–> You may be asked to synthesize a product given a set of vague or specific reaction conditions.
For example:
“Synthesize 2-butanone using any inorganic reagents. Use 2 carbon alkyl halides as your only carbon source.”
Have you seen similar questions in your homework, quizzes or practice exams?
What do you think?
Many students will look at the process and panic;
And in said panic started drawing everything and anything that comes to mind, without a clear process or idea of where they are headed.
I like to be systematic in my approach to problems.
I like to plan my steps and know exactly what I have to do.
And more importantly, I trust that the process will help me get the correct results, again and again.
As quickly as possible!
The more systematic your approach, the more likely you’ll get every component of the reaction. Therefore, the less likely you’ll miss something.
That’s the key. Get every component and minimize lost points on your exam.
Before even diving into the problems themselves, you need to know where you’re going.
You must ensure that what you do will ultimately pay off to give you your desired product.
These are my go-to Retrosynthesis questions:
- What’s the same?
- What’s different?
- How can I achieve this difference?
What’s the Same?
Before adding new groups to the molecule, you want to see what is already there to work with.
We’ll utilize the analysis taught in the synthesis tutorial when analyzing what’s present on the reactant.
COUNT YOUR CARBONS!!!
(so many lose points due to lack of counting.)
How many carbon atoms are present in the reactant and product?
This will help you identify chain elongation or cleavage reactions.
Look for any non-carbon atoms and functional groups.
What else is on the molecule in the reactant and product?
Look for location of reactivity on the molecule.
Reactivity on the molecule refers to the location of reactive atoms or functional groups.
For example, 2-chloropropane has reactivity on the second carbon. Cl is a good leaving group. This allows us to carry out a number of reactions:
- Elimination to create a pi bond (E1 or E2)
- Substitution to give us another functional group in the form of an incoming nucleophile (SN1 or SN2)
- We could turn the Cl into a Grignard for a super reactive organometallic
- And so much more…
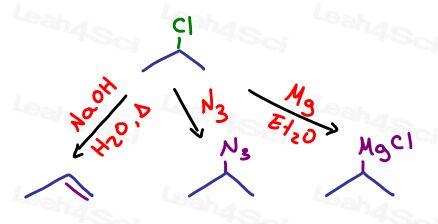
This is your clue regarding WHERE to start the reaction.
Compare each of these features to the final product.
Make careful note of anything that changes because our goal in carrying out retrosynthesis will be exactly that: figuring out HOW to carry out these transformations, which brings me to question 2.
What’s Different?
- If the number of carbon atoms changed, by how many?
- How many carbons were added or removed?
- If the functional groups or reactivity changed, what was replaced and by what?
- How many new groups were added?
Once we’ve identified what’s the same and what’s different, we ask the most important question:
HOW Can I Carry Out this Transformation?
This is a two-part question.
1) For short synthesis problems the answer is simple.
Which ONE reaction will convert starting molecule x to end with product y?
For example, 2-iodopropane to propene.
What’s the same?
Both reactant and product have three carbon atoms.
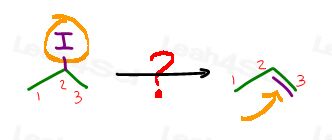
What’s different?
The reactant has a halogen at carbon #2.
The product has a pi bond between former carbon 2 and carbon 3.
How can I carry out this transformation?
Eliminate the halogen using a strong base for an E2 reaction or even a weak base and heat for an E1 reaction. Both will provide the same product.
Quick tip: When in doubt select E2 over E1. It’s faster, more precise and has less competition (E1 vs SN1) when conditions are set right. You can also control the product choosing to form a more (Zaitsev) or less substituted (Big Bulky Base) pi bond.
2) But your professor will likely not give you such a simple one-step transformation for retrosynthesis.
Let’s crank it up a notch.
For example, say you’re asked to convert 2-chlorobutane to 1-butanol.
Still a relatively simple transformation, but we’re no longer looking at a simple one-step reaction.
The same set of questions apply and will still guide you to the product.
What’s the same?
We have a total of four carbons in the reactant and product.
We have a single functional group in the reactant and product.
What’s different?
The reactant has a halogen; the product has an alcohol.
Reactivity on the molecule shifted from carbon #2 to carbon #1.
Which reactions do I know to carry out this transformation?
Suddenly the answer is not as clear, but it is still not impossible.
In the previous example we were able to quickly answer this question.
ONE single reaction transformed our starting molecule to our desired product.
Here, not so much!
This is where the true power of retrosynthetic analysis comes into play.
Let’s break it down.
We have a chlorine on the starting molecule and alcohol on the product.
While we DO know of a reaction to convert Cl to OH (SN2) this will happen on the same carbon. We need the OH on a different carbon.
So let’s take it a step further or should I say take it a step back.
Retrosynthesis IS backwards thinking, so let’s start with the product.
If the product is an alcohol on the primary carbon, what reaction do I know that will GIVE ME an alcohol on the primary carbon?
Primary vs secondary tells me Anti-Markovnikov alcohol, which tells me I need to carry out an alkene addition reaction under Anti-Markovnikov conditions.
Taking the product just one step back, I need an alkene.
Don’t worry about the reagents just yet. It’s much easier to think through the molecules first and then go back and fill in the missing reagents as explained in the synthesis tutorial.
Now we treat the alkene as our new product and ask the same question again.
Comparing the reactant to the alkene,
Do I know of a reaction that will either carry out this transformation or get me close?
Why absolutely yes, a simple elimination reaction as we’ve seen above.
However, this time we can’t simply use a strong base because the pi bond will favor the more substituted and Zaitsev product 2-butene.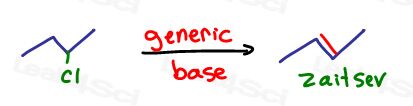
Instead, we need an elimination reaction that will force the pi bond to form on the less substituted primary to secondary carbon.
Which reagent will carry out this ‘anti-Zaitsev’ or Hoffman elimination?
We need a ‘triple B’ or Big Bulky Base
Tert-butoxide
But let’s not worry about tert-butoxide right now, instead let’s simply acknowledge the fact that we CAN react 2-chlorobutane to form 1-butene and draw this transformation.
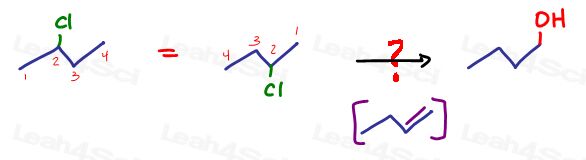
Once you have all of your intermediates drawn in from product to reactant, quickly follow the sequence from reactant to product to ensure it looks right and makes sense.

Now ask yourself this:
Which reagent will carry out each transformation?
Ask yourself this question one at a time as you fill in the reaction conditions and complete your retrosynthesis sequence.
We start with a secondary halogen and form a less substituted pi bond.
This requires the strong base tert-butoxide as we already hinted above.
While some professors will accept this as is, others will require a full set of conditions.
Let’s use potassium tert-butoxide dissolved in it’s own conjugate alcohol tert butanol and heat to help carry out this reaction. Recall that E2 reactions prefer heat. Now that we have an asymmetric alkene we need an alkene addition reaction that will allow an alcohol to add onto a primary carbon or the anti-Markovnikov position.
Now that we have an asymmetric alkene we need an alkene addition reaction that will allow an alcohol to add onto a primary carbon or the anti-Markovnikov position.
Hydroboration oxidation should immediately come to mind.
Let’s add in BH3, THF in NaOH and H2O2.
And there we have it, a step by step transformation with reagents in place.
Retrosynthesis is Really a Combination of Forward and Reverse Thinking.
Synthesis + retrosynthetic analysis.
Start with backwards thinking whenever you can.
You know the expression “hindsight is 20/20?”
That’s what we’re banking on.
By looking at your product and only keeping your reactant in mind, you’re able to ask the very, very important question:
What did this molecule look like just ONE step prior, so that I can form functional group x?
This form of retrosynthetic analysis will help you quickly identify one intermediate at a time, all the way back to your starting molecule.
Let’s try something a little more complex.
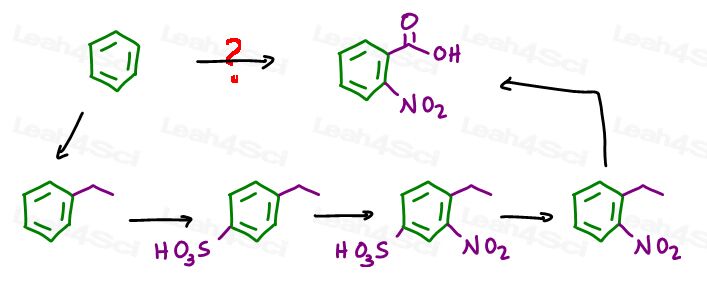
Say you’re asked to start with benzene to synthesize 2-nitrobenzoic acid.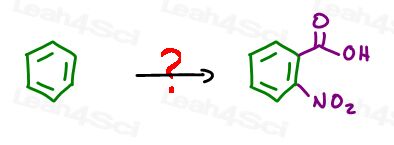
What’s the same?
Reactant and product have a benzene ring.
What’s different?
The product is a disubstituted benzene. The substituents have an ortho relationship.
We’ve added a carboxylic acid and a nitro group.
Which reactions do I know to carry out these transformations?
Given that we have more than one reaction taking place, always pay attention to how the reactions impact each other. 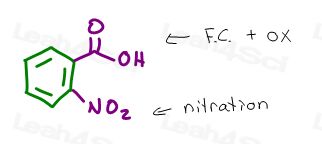
We can add the carboxylic acid through a Friedel Crafts Alkylation followed by oxidation.
We can add the nitro group through EAS Nitration.
BUT, here’s what you want to be careful about: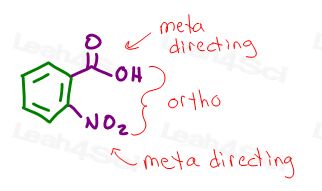
The groups are ortho to each other.
Both NO2 and carboxylic acid are EAS deactivating groups with meta directing effects.
Let’s think this through before we start reacting.
If the carboxylic acid comes from an FC Alkylation, the alkyl group prior to oxidation is an ortho/para director.
That’s good.
But how do we ensure that we wind up with the ortho rather than para position?
We need one more reaction that is not apparent in the product.
We need a blocking group at the para position to ensure ortho is the only available group.
All these thoughts should quickly run through your head. There’s no need to write the reactions just yet, although I do like to mark up the molecules as a ‘note to self.’
Now that we know where we’re going, retrosynthesis is done and we can take this reaction from reactant to product directly.
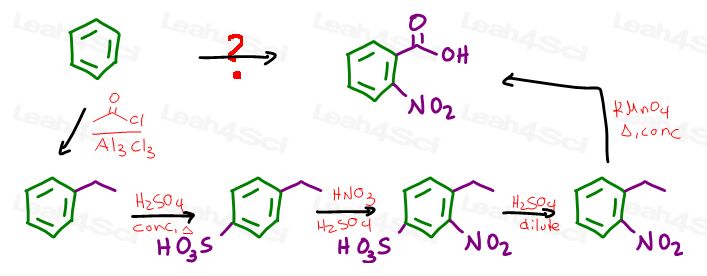
We’ll start with a Friedel Crafts alkylation. The size of your carbon chain doesn’t matter since side chain oxidation will cut off the extra carbon atoms.
Let’s go with an ethyl chloride in FeCl3 giving us ethylbenzene: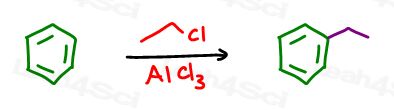
We now have an ortho para director, so let’s add the blocking group.
EAS sulfonation will add the bulky SO3H at the para position.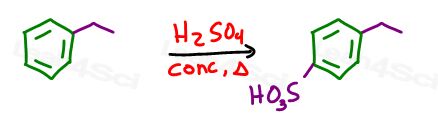
With para blocked we can carry out another reaction keeping in mind the following:
- Ethyl is still an ortho/para director but para is blocked!
- Sulfur is partially positive and a meta directing group.
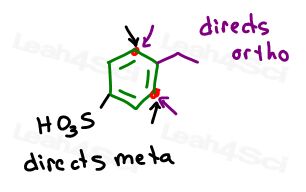
BOTH groups direct to the #2 carbon near the ethyl group which is exactly what we want.
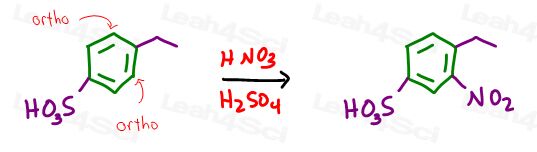
Next we carry out EAS nitration forcing the nitro group ortho to the ethyl and meta to the sulfate.
Dilute acid will then remove the blocking group from the para position, leaving you with a nitro group meta to the ethyl group. 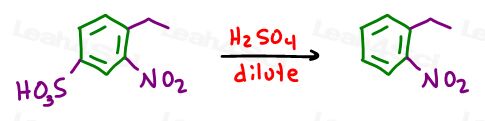
This is where many students lose points.
They get so excited for having thought of all this that they forget that this is not the desired product.
ALWAYS go back to the initial question to ensure you haven't forgotten anything.
What have we forgotten?
Ethyl is not our goal.
We need a carboxylic acid.
Enter potassium permanganate with is side chain oxidation powers turning the benzylic position into a carboxylic acid for our final product.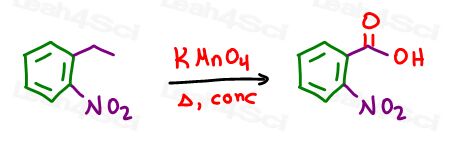
Once done, run through the entire sequence to ensure all your reagents and intermediates work together and make sense and we’re done.
 These same thoughts can be applied to any retrosynthesis problem from two through 100 steps and more. Ok, perhaps I’m exaggerating, your professor will hopefully limit your retrosynthesis problems to three to seven steps.
These same thoughts can be applied to any retrosynthesis problem from two through 100 steps and more. Ok, perhaps I’m exaggerating, your professor will hopefully limit your retrosynthesis problems to three to seven steps.
Now that you have the basics for how to approach retrosynthesis, you will need a solid foundation. I recommend going back to review all the key reactions covered in your semester so you have them fresh and ready to utilize as needed.
Use my Syllabus Companion to quickly find tutorials, videos and cheat sheets for typical organic chemistry reactions.
I’d love to hear from you
Do you feel better about retrosynthesis and staying on the right path to full credit for each problem? Let me know in the comments below.